- · 《海外文摘·学术版》栏目[05/29]
- · 《海外文摘·学术版》刊物[05/29]
- · 《海外文摘·学术版》征稿[05/29]
- · 《海外文摘·学术版》投稿[05/29]
- · 《海外文摘·学术版》收稿[05/29]
一、来稿必须是作者独立取得的原创性学术研究成果,来稿的文字复制比(相似度或重复率)必须低于用稿标准,引用部分文字的要在参考文献中注明;署名和作者单位无误,未曾以任何形式用任何文种在国内外公开发表过;未一稿多投。 二、来稿除文中特别加以标注和致谢之外,不侵犯任何版权或损害第三方的任何其他权利。如果20天后未收到本刊的录用通知,可自行处理(双方另有约定的除外)。 三、来稿经审阅通过,编辑部会将修改意见反馈给您,您应在收到通知7天内提交修改稿。作者享有引用和复制该文的权利及著作权法的其它权利。 四、一般来说,4500字(电脑WORD统计,图表另计)以下的文章,不能说清问题,很难保证学术质量,本刊恕不受理。 五、论文格式及要素:标题、作者、工作单位全称(院系处室)、摘要、关键词、正文、注释、参考文献(遵从国家标准:GB\T7714-2005,点击查看参考文献格式示例)、作者简介(100字内)、联系方式(通信地址、邮编、电话、电子信箱)。 六、处理流程:(1) 通过电子邮件将稿件发到我刊唯一投稿信箱(2)我刊初审周期为2-3个工作日,请在投稿3天后查看您的邮箱,收阅我们的审稿回复或用稿通知;若30天内没有收到我们的回复,稿件可自行处理。(3)按用稿通知上的要求办理相关手续后,稿件将进入出版程序。(4) 杂志出刊后,我们会按照您提供的地址免费奉寄样刊。 七、凡向文教资料杂志社投稿者均被视为接受如下声明:(1)稿件必须是作者本人独立完成的,属原创作品(包括翻译),杜绝抄袭行为,严禁学术腐败现象,严格学术不端检测,如发现系抄袭作品并由此引起的一切责任均由作者本人承担,本刊不承担任何民事连带责任。(2)本刊发表的所有文章,除另有说明外,只代表作者本人的观点,不代表本刊观点。由此引发的任何纠纷和争议本刊不受任何牵连。(3)本刊拥有自主编辑权,但仅限于不违背作者原意的技术性调整。如必须进行重大改动的,编辑部有义务告知作者,或由作者授权编辑修改,或提出意见由作者自己修改。(4)作品在《文教资料》发表后,作者同意其电子版同时发布在文教资料杂志社官方网上。(5)作者同意将其拥有的对其论文的汇编权、翻译权、印刷版和电子版的复制权、网络传播权、发行权等权利在世界范围内无限期转让给《文教资料》杂志社。本刊在与国内外文献数据库或检索系统进行交流合作时,不再征询作者意见,并且不再支付稿酬。 九、特别欢迎用电子文档投稿,或邮寄编辑部,勿邮寄私人,以免延误稿件处理时间。
周末文摘 | 细胞和基因治疗产品监管政策的中美
作者:网站采编关键词:
摘要:引用本文 王刚.细胞和基因治疗产品监管政策的中美比较[J].中国食品药品监管.2019.8(187):20-25. 细胞和基因治疗是当前备受 关注 的创新技术。细胞治疗产品包括细胞免疫疗法、癌症疫


引用本文
王刚.细胞和基因治疗产品监管政策的中美比较[J].中国食品药品监管.2019.8(187):20-25.
细胞和基因治疗是当前备受关注的创新技术。细胞治疗产品包括细胞免疫疗法、癌症疫苗和用于某些适应证的其他类型的自体和同种异体细胞,包括造血干细胞、成体和胚胎干细胞。基因疗法试图修饰或操纵基因的表达或改变活细胞的生物学特性,以用于治疗人体疾病。
截至2018 年底,美国已有逾800 项与细胞和基因治疗相关的临床研究正在进行。为促进国内细胞和基因治疗产业发展,本文将从产品和监管两个方面概述FDA 对细胞治疗产品监管的相关要求,探讨细胞和基因治疗领域监管政策。
2018 年末,FDA 时任局长Scott Gottlieb提出,预计到2020 年,FDA 每年将收到逾200项细胞治疗和基因治疗产品的新药临床试验(即IND)注册申请。到2025 年,FDA 每年将批准10~20 项细胞和基因治疗产品上市销售。为了应对新技术快速增长带来的巨大挑战,未来几年,FDA 将在增加常规审评员、监管人员的基础上,再增加50 位临床审评员,定向解决临床方面的审评需求。此外,FDA 还将进一步出台一系列相关政策支持细胞和基因治疗产品的研制和申报,针对加速审评审批建立新机制。
鼓励政策
在美国,FDA 的生物制品评价与研究中心(CBER,Center for Biologics Evaluationand Research)主要负责生物制品包括细胞治疗和基因治疗产品的审评。该部门的常规业务是传统生物大分子药物如疫苗、血液产品及细胞治疗和组织等产品的审评和监管。由于当前基因和细胞治疗产品备受关注,其审评和监管的需求量也在显著增加。
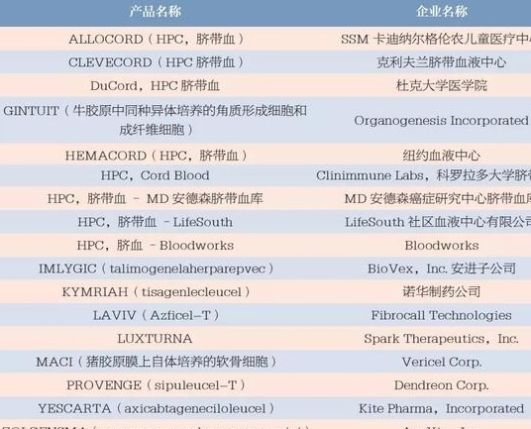
1997 年, 第一款由健赞公司生产的用于修复大腿关节软骨的细胞治疗产品Carticel 被FDA 批准上市,但真正意义上的经过严格临床试验和审批的细胞治疗产品出现在2010 年,即Dendreon 生产的用于治疗前列腺癌的细胞免疫产品Provenge。过去十几年中,FDA 批准了为数不多的细胞和基因治疗产品(表1)。行业研发热情高涨,也是受到了产品获批,特别是2015年批准的溶瘤病毒产品、2017 年两款CAR-T 产品的影响。
与美国近千项细胞和基因治疗相关研究相比,中国国家药品监督管理局药品审评中心(以下简称CDE)在过去几年中只批准了近20 项细胞治疗产品的IND 临床研究,直到最近才批准了第一个基因治疗产品的IND,国内外数量差距很大,以此推测,今后上市产品数量差距会更加凸显。究其原因,这种差距与2016 年发生的“魏则西事件”所带来的负面影响不无关系,但与彼时政策相对滞后和保守及监管环境的不明确也有很大关系。
由于IND 审评速度的滞后及中国在细胞治疗产品监管方面所采纳的“双轨制”,即CDE 的药品“注册制”和卫健委的医疗技术“备案制”,很多国内企业和研究机构通过要求相对较低的“备案制”在医疗机构开展临床研究,而非通过要求相对严格的“注册制”开展IND 研究。这种做法虽然在一定程度上推动了细胞治疗产品和行业在临床应用方面的发展,但在很大程度上阻碍了细胞治疗产品按药品研发和申报途径向药监部门提交IND 申请,从而导致了一种奇怪现象,即中国在美国国家医学图书馆和美国国家卫生研究院联合开办的官方网站上(www.)登记注册的在中国开展的CAR-T 临床研究项目多达250 多个,不比美国正在开展的CAR-T 临床研究少多少,但真正在药监部门注册的IND 数量却要少得多。

2017 年FDA 分别批准了诺华和凯特制药的两款CAR-T 产品,即KYMRIAH 和YESCARTA ;同年12 月22 日,CDE 出台《细胞治疗产品研究与评价技术指导原则》,表明审评部门对此类产品的了解愈加深入,监管途径日趋清晰,从而显著促进了中国企业从“幕后”研究走向“台前”。与此同时,一些国外著名药企也纷纷与国内企业合作,进一步推动细胞治疗产品的发展。2017 年底,一大批自主研发的CAR-T 药物开始申报,旨在通过“注册制”渠道成为药品。
文章来源:《海外文摘·学术版》 网址: http://www.hwwzzz.cn/zonghexinwen/2020/0709/356.html
上一篇:学术文摘丨论聚落遗产与价值体系的建构(上)
下一篇:文艺在线·夜听|诗朗诵:黄梅戏